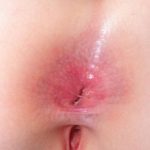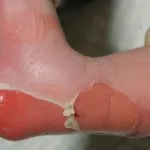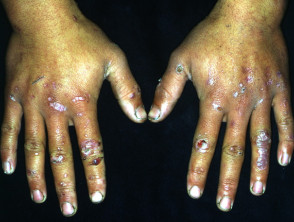
¿Qué es la epidermólisis ampollosa?
Epidermólisis ampollosa (EB) es el nombre que se le da a un grupo de enfermedades ampollosas hereditarias que están presentes desde el nacimiento.
¿Qué es la epidermólisis ampollosa adquirida?
Epidermólisis ampollosa adquirida (EBA) es un raro autoinmune enfermedad con ampollas en el que aparecen ampollas subepiteliales tensas en los sitios de trauma. A diferencia de la EB, la ABE no se hereda y generalmente se presenta en la vida adulta.
Las ampollas de EBA tienden a ser localizado a áreas que se lesionan fácilmente, como las manos, los pies, las rodillas, los codos y las nalgas. A veces hay mucosa afectación con formación de ampollas en la boca, nariz y ojos.
Epidermólisis ampollosa adquirida
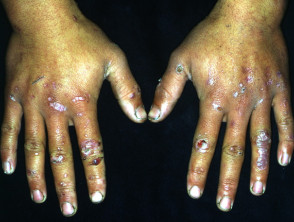
Epidermólisis ampollosa adquirida
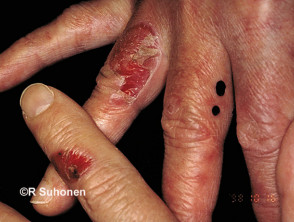
Epidermólisis ampollosa adquirida
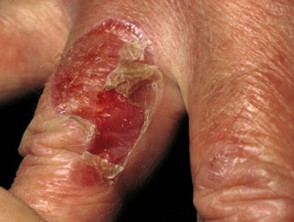
Epidermólisis ampollosa adquirida
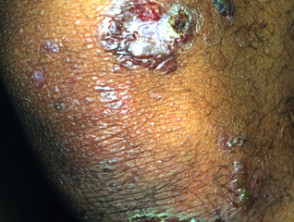
Epidermólisis ampollosa adquirida
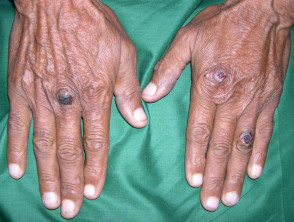
Epidermólisis ampollosa adquirida
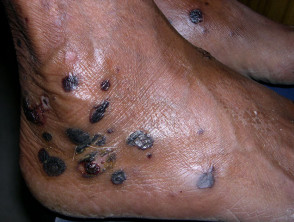
Epidermólisis ampollosa adquirida
¿Quién contrae la epidermólisis ampollosa adquirida y por qué?
La ABE generalmente ocurre en la cuarta y quinta décadas de la vida. Pueden verse afectados machos y hembras de todas las razas.
Se ha informado que algunos pacientes con ABE tienen otras enfermedades autoinmunes, con mayor frecuencia enfermedad de Crohn y sistémico lupus eritematoso, o problemas de salud como amilosis, mieloma múltiple y, raramente, carcinoma del pulmón y linfoma. Otros pacientes solo tienen un problema de piel.
¿Cuál es la causa de la epidermólisis ampollosa adquirida?
Los investigadores han detectado tejidos unidos inmunoglobulina (Ig) -G autoanticuerpos dirigido contra el dominio NC1 de tipo VII colágeno (Col7) en pacientes afectados. El colágeno tipo VII es el principal componente estructural del anclaje. fibrillas. Estos adjuntan el membrana basal del epidermis al subyacente dermis. La pérdida de fibrillas de anclaje conduce a la formación de ampollas justo debajo de la epidermis dentro de un área conocida como lámina densa.
¿Cuáles son las características clínicas de la epidermólisis ampollosa adquirida?
EBA tiene varias presentaciones clínicas distintas.
| Clasificación de EBA | Características clínicas |
|---|---|
|
Forma clásica mecanobullosa de EBA |
|
|
Forma no clásica / no mecanobullosa de EBA |
|
|
Predominantemente membrana mucosa forma de EBA |
|
|
ABE tipo Brunsting-Perry |
|
|
IgA ‐ EBA |
|
¿Cómo se diagnostica la epidermólisis ampollosa adquirida?
Se deben realizar las siguientes pruebas para establecer un diagnóstico de EBA.
- Piel biopsia - esto muestra la presencia de una ampolla debajo de la epidermis (subepidérmico bula); una variable infiltrado de mixto inflamatorio células de la dermis; y quistes milia y cicatrices en lesiones más antiguas.
- Directo inmunofluorescencia (DIF) de una biopsia de piel - esto muestra declaración de inmunoglobulinaIgG con más frecuencia que IgM o IgA) y C3 en una línea a lo largo de la unión dermoepidérmica. Vasos sanguineos no están involucrados.
- Inmunofluorescencia indirecta (IIF) de suero - esto revela bajo titulo autoanticuerpos IgG circulantes que se dirigen a la membrana basal.
Otras pruebas para confirmar el diagnóstico solo están disponibles en algunos centros académicos. Incluyen [2]:
- Biopsia de piel para inmunoelectrones directos e indirectos microscopía, análisis de patrón de dentado por DIF, superposición fluorescente antígeno mapeo y DIF en piel dividida por sal
- Análisis de sangre para detectar circulante anticuerpos a Col7: inmunotransferencia, enzimaensayo inmunoabsorbente ligado (ELISA), IIF más detallado e inmunoprecipitación.
El Grupo Internacional de Enfermedades Bullosas (IBDG) ha propuesto por consenso criterios clínicos y diagnósticos para la ABE [1,2]. Si una enfermedad con ampollas es clínica y histológicamente compatible con EBA, el diagnóstico se confirma si:
- La deposición lineal de IgG y / o C3 a lo largo de la zona de la membrana basal se demuestra mediante inmunofluorescencia directa (DIF) y,
- Se detectan autoanticuerpos contra COL7 en el suero del paciente (mediante ensayo inmunoabsorbente ligado a enzimas).
Cuál es el diagnóstico diferencial para la epidermólisis ampollosa adquirida?
EBA puede imitar otras enfermedades inflamatorias ampollosas, con mayor frecuencia penfigoide ampolloso y lupus eritematoso sistémico ampolloso (que también se asocia con autoanticuerpos anti-Col7).
En comparación con el penfigoide ampolloso, EBA [2]:
- Surge en personas más jóvenes, generalmente menores de 70 años
- Tiene una participación prominente de la cabeza y el cuello.
- Afecta a las membranas mucosas
- Las lesiones se curan con cicatrices y formación de milia.
¿Cuál es el tratamiento para la epidermólisis ampollosa adquirida?
los primario El objetivo del tratamiento de EBA es proteger la piel y detener la formación de ampollas, promover la cicatrización y prevenir complicaciones.
Dado que la ABE se considera una enfermedad autoinmune, es razonable su uso inmunosupresor agentes para modificar o reducir las respuestas autoinmunes y disminuir la producción de autoanticuerpos. Estos pueden incluir:
- Azatioprina
- Dapsona
- Colchicina
- Corticosteroides
- Micofenolato de mofetilo
- Oro
- Inmunoglobulina intravenosa.
Debido a la rareza de la enfermedad, es difícil estar seguro de qué fármaco es el más eficaz.
Otras estrategias de gestión importantes incluyen:
- Tratamiento de cualquier enfermedad subyacente.
- Consulta con otros especialistas como dentista y oftalmólogo.
- Evite el trauma físico directo a las superficies de la piel.
- Evite los alimentos duros o quebradizos o los alimentos con alto contenido de ácido en pacientes con afectación bucal.
Cuál es el pronóstico?
EBA es una enfermedad inflamatoria crónica que tiene períodos de parcial remisiones y exacerbaciones. Si se trata y se cuida adecuadamente, los pacientes pueden esperar vivir una vida normal. Los problemas que pueden surgir incluyen:
- Infección de heridas abiertas y llagas
- Cicatrices severas que pueden causar deformidades y restricción de movimiento
- Enfermedad periodontal
- Cicatrices conjuntivales y ceguera
- Efectos secundarios de la medicación