¿Qué es la deficiencia del interleucina-1 receptor antagonista (DIRA)?
La deficiencia del antagonista del receptor de interleucina-1 (DIRA) (MIM 612852) es una genético autoinflamatorio síndrome que se presenta en los primeros días de vida. El severo inflamatorio La reacción se asemeja a una agudo grave sistémico infección o infección del hueso. Si se reconoce temprano, el tratamiento con anakinra, un agente biológico, puede prevenir la muerte por falla multiorgánica.
¿Quién recibe DIRA y por qué?
La DIRA se ha diagnosticado solo en un número muy reducido de niños. Se presenta al nacer o en los primeros días de vida. Se han identificado casos de familias originarias de Puerto Rico, Terranova (Canadá), Países Bajos y Líbano.
DIRA es una autosómico trastorno genético recesivo, es decir, cada hijo de dos portador los padres tienen un 25% de posibilidades de sufrir DIRA.
Molecular biología y genética
DIRA se debe a homocigoto punto mutaciones en el gene IL1RN (2q14.2) o una gran deleción genómica que afecta a este gen y a otros de la familia IL en cromosoma 2. Lo mismo mutación se detecta en todos los pacientes que se originan en un área determinada, lo que sugiere un efecto fundador distinto en cada región. Estudios de normal control S de las mismas áreas encontraron tasas de portadores para la mutación de 0.2% en Terranova y 1.3% en Puerto Rica. Estas mutaciones, cuando son homocigotas, dan como resultado la ausencia completa del antagonista funcional del receptor de interleucina-1. proteína. Por tanto, la interleucina-1 tiene efectos sin oposición sobre el receptor. La estimulación del receptor de interleucina-1 es un poderoso activador de las vías inflamatorias.
Características clínicas de DIRA
Las características distintivas de este síndrome están presentes al nacer, o pocos días después del nacimiento, y se deben a cambios inflamatorios en la piel y los huesos en ausencia de fiebre.
En los primeros días de vida, las características de presentación comunes incluyen:
- Sufrimiento fetal
- Pustuloso erupción
- Boca lesiones
- Inflamación de articulaciones
- Movimiento doloroso
- Agrandamiento del hígado y el bazo (hepatoesplenomegalia)
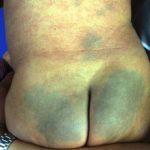
Todos los niños afectados desarrollar la erupción pustulosa, en la que puede haber discreto cultivos de pústulas o un severo generalizado pustuloso erupción que puede parecerse a la psoriasis pustulosa generalizada.
Otra piel /mucosa los cambios informados incluyeron:
- Generalizado ictiosis-como cambios
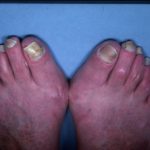
- Uña cambios - picadura, separación de la uña del lecho ungueal, como psoriasis ungueal
- Estomatitis - puede ampollarse
- Boca úlceras
- Conjuntivitis (dolor en los ojos)
- Pioderma gangrenoso (un caso)
Todos los niños se desarrollan inflamación en los huesos, que provocan movimientos dolorosos e inflamación de las articulaciones. Los cambios en las radiografías son característicos.
Otras características observadas en casos individuales incluyen:
- Inflamación de vasos sanguineos en el cerebro (cerebral vasculitis)
- Pulmonar hemosiderosis (hierro declaración en los pulmones) con progresivo intersticial fibrosis (cicatrices)
- Hipotonía (tono muscular débil)
- Fracaso para prosperar
La fiebre alta no es una característica típica.
Si no se trata, DIRA produce insuficiencia multiorgánica y muerte en la infancia.
¿Cómo se diagnostica DIRA?
Las características de presentación de DIRA sugieren una infección sistémica aguda grave (neonatal septicemia) y / o infección del hueso (osteomielitis); sin embargo, todas las investigaciones de infección son negativas.
La siguiente tabla describe los resultados de las investigaciones.
| Análisis de sangre |
|
| Piel biopsia |
|
| Rayos X |
|
| Biopsia de hueso |
|
| Cerebral Resonancia magnética |
|
| ADN análisis |
|
Tratamiento de DIRA
Los niños con DIRA pueden tratarse eficazmente con anakinra, un agente biológico que es una forma recombinante de antagonista del receptor de interleucina-1, la proteína de la que carecen estos niños. Recientemente se ha puesto a disposición para tratar la enfermedad reumatoide artritis cuando DIRA se describió y caracterizó por primera vez en detalle.
Diario subcutáneo La administración de anakinra reemplaza la proteína faltante con una excelente respuesta, particularmente en aquellos niños con mutaciones puntuales. En la serie de casos inicial reportada, ocurrió una resolución completa y rápida de los síntomas y signos. Los cambios en la piel respondieron en unos días y las radiografías de los huesos se normalizaron en meses. Los marcadores sanguíneos de inflamación también volvieron a niveles normales. La respuesta en los niños con la deleción genómica grande fue menos dramática ya que los marcadores sanguíneos no se normalizaron. La dosis de anakinra se inició con 1 mg / kg / día y se incrementó en 0,5 mg / kg / día hasta que se logró la respuesta. Se ha informado que la resolución de los síntomas y signos es duradera, con un rápido deterioro dentro de las 36 horas posteriores a la interrupción del tratamiento con anakinra. Por tanto, el tratamiento será de por vida. Las reacciones locales en el lugar de la inyección (enrojecimiento, picazón, hinchazón) son un efecto adverso común, pero por lo demás el tratamiento se tolera bien. El seguimiento a largo plazo aún no está disponible.
Nota: anakinra no está registrada ni subvencionada en Nueva Zelanda. En otros países como EE.UU. y Europa, su indicación registrada es la artritis reumatoide.