¿Qué es la hiper-IgM ligada al cromosoma X? síndrome?
El síndrome de hiper-IgM ligado al cromosoma X (OMIM 308230) es un genético síndrome de inmunodeficiencia que se presenta con bacteriano y oportunista infecciones desde una temprana edad. Es la forma más común de síndrome de hiper-IgM y representa aproximadamente el 70% de los casos. Un nombre antiguo para esta enfermedad era disgammaglobulinemia tipo I con neutropenia.
¿Quién contrae el síndrome de hiper-IgM ligado al cromosoma X y por qué?
El síndrome de hiper-IgM ligado al cromosoma X es una condición rara, con un estimado incidencia de aproximadamente 1 caso por millón de habitantes.
El síndrome de hiper-IgM ligado al cromosoma X solo afecta a los hombres, ya que el gene se encuentra en la X cromosoma. Se hereda de la madre que es asintomático portador. El diagnóstico generalmente se realiza antes de los 12 meses cuando el bebé varón presenta infecciones inusuales o graves. Puede haber antecedentes familiares de muertes inexplicables en bebés varones.
El defecto genético involucra al gen en el cromosoma Xq26 que codifica el ligando CD40. Esta molécula se expresa en CD4 + activadoT linfocitos y participa en el cambio Células B de hacer IgM anticuerpos a IgG y otras formas de anticuerpo. Entonces, aunque el problema se presenta como una incapacidad de las células B para producir una respuesta de anticuerpos adecuada a las infecciones, el defecto está en Células T.
Características clínicas
Esta afección generalmente se presenta como infecciones inusuales, oportunistas o graves de las vías respiratorias superiores e inferiores o del tracto gastrointestinal y a menudo se asocia con retraso del crecimiento y agrandamiento linfa nodos.
- La neumonía afecta a más del 80% de los pacientes; La neumonía por Pneumocystis jiroveci es el problema de presentación en el 40% (anteriormente conocido como Pneumocystis carinii)
- Infecciones de oído (otitis)
- Seno infeccionessinusitis)
- Infecciones del sistema nervioso central (infecciones bacterianas, virales y micóticas)
- Invasor las infecciones por hongos incluyen cándida, histoplasmosis, criptococosis
- Crónico La diarrea debida a la ciptosporidiosis puede provocar colangitis esclerosante e hígado. cirrosis
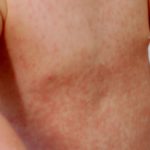
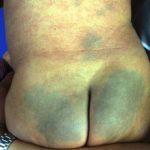
Los signos dermatológicos incluyen frecuentes y recurrente Infecciones bacterianas y micóticas de la piel y oral. úlceras.
Sistémico infección con piel manifestaciones incluyen criptococosis e histoplasmosis. Primario La infección de la piel puede ser bacteriana, incluida la celulitis, o viral, incluidas las verrugas resistentes al tratamiento, el herpes labial (herpes simplex)
Las úlceras orales no suelen deberse a infecciones, aunque deben excluirse. Las características de las úlceras incluyen:
- recurrente, grande (hasta 2 cm)
- puede ser múltiple
- doloroso lesiones puede requerir alivio del dolor
- generalmente afectan la lengua y el interior de las mejillas
- normalmente cura sin dejar cicatrices
- comienzan en la primera infancia, comúnmente en el primer año
- tarda en sanar: de 3 semanas a 3 meses
- puede interferir con la alimentación / alimentación y la higiene bucal
- a veces se relacionan con episodios de neutropenia
Otras características de esta condición incluyen:
- Gingivitis - estreptocócica
- Estomatitis (oral inflamado mucosa)
- Trombocitopénico púrpura (sangrado debido a baja plaqueta contar)
- Neutropenia que puede ser intermitente o persistente
- Autoinmune condiciones que incluyen Coombs positivo anemia, trombocitopenia, inflamatorio enfermedad intestinal, hepatitis
- Cáncer - del biliar árbol e intestino
¿Cómo se diagnostica el síndrome de hiper-IgM ligado al cromosoma X?
El diagnóstico temprano es importante ya que el síndrome de hiper-IgM ligado al cromosoma X no tratado es fatal.
Las investigaciones iniciales para un paciente sospechoso de tener un síndrome de inmunodeficiencia incluyen inmunoglobulina (anticuerpos) en la sangre.
En el síndrome de hiper-IgM ligado al cromosoma X:
- IgG: muy baja o indetectable
- IgE: muy baja o indetectable
- IgA: generalmente muy baja, pero puede ser normal o alta
- IgM: generalmente alto, aunque también se han informado niveles normales o bajos.
- Tal resultado de análisis de sangre sugeriría el diagnóstico de un síndrome de hiper-IgM.
Las investigaciones específicas para el síndrome de hiper-IgM ligado al cromosoma X son:
- La citometría de flujo de linfocitos T activados no detectará el ligando CD40
- ADN Se debe realizar la secuenciación del gen específico en el cromosoma X para identificar la mutación y confirmar el diagnóstico. Muchas diferentes mutaciones se han identificado en este gen.
Estas pruebas distinguirán el síndrome de hiper-IgM ligado al cromosoma X del más raro autosómico síndrome de deficiencia de CD40 recesivo, que es clínicamente idéntico. Además, ataxia-telangiectasia puede presentarse como un síndrome de hiper-IgM en la infancia con el neurológico síntomas y telangiectasia desarrollando más tarde en la niñez. Congénito La rubéola, el cáncer y los medicamentos antieplilépticos también pueden causar un patrón de inmunodeficiencia hiper-IgM.
El diagnóstico del estado de portador debe realizarse en ambos padres, esperando que la madre tenga una copia de la misma mutación genética (y un gen normal) y que el padre tenga un gen normal. Prenatal el diagnóstico está disponible.
Tratamiento del síndrome de hiper-IgM ligado al cromosoma X
La gammaglobulina intravenosa mensual debe iniciarse tan pronto como se haga el diagnóstico, ya que esto produce niveles normales de IgM y limita el número de infecciones graves y graves. Sin embargo, es posible que no prevenga las infecciones oportunistas. En algunos casos también mejora el crecimiento.
Trimetoprim + sulfametoxazol puede ser dado profilácticamente para prevenir la infección por Pneumocystis jirovecii.
Granulocito El factor estimulante de colonias (GCSF) trata la neutropenia.
Deben evitarse las vacunas vivas.
Médula ósea o umbilical cable célula madre trasplante ha sido curativo en algunos pacientes.
Se ha sugerido que los niños afectados deben recibir inicialmente gammaglobulina intravenosa y trimetoprima + sulfametoxazol con un seguimiento cuidadoso de las complicaciones de la enfermedad. Médula ósea trasplante debe ser considerado y realizado si es posible cuando las complicaciones desarrollar.