Comprensión del Síndrome de Microftalmia con Defectos Lineales de la Piel (MLS)
¿Qué es el Síndrome de Microftalmia con Defectos Lineales de la Piel?
El síndrome de microftalmia con defectos lineales de la piel (MLS), identificado con el código MIM309801, es un trastorno del neurodesarrollo de origen genético que se manifiesta al nacer, afectando predominantemente a mujeres. Sus dos manifestaciones clínicas primordiales involucran anormalidades en los ojos y en la piel, aunque frecuentemente se asocian también con complicaciones neurológicas y cardiovasculares.
El síndrome MLS recibe diversas denominaciones alternativas, incluyendo síndrome MIDAS (Microftalmia, Aplasia Dérmica, Esclerocórnea) o el síndrome de Gazali-Temple.
¿Quién Desarrolla el Síndrome MLS y Cuál es su Etiología?
El síndrome MLS se clasifica como una condición genética dominante ligada al cromosoma X, lo que explica su predominio en el sexo femenino. Se ha documentado su transmisión directa de madre a hija. La inactivación del cromosoma X juega un papel crucial en la modulación de las manifestaciones clínicas del síndrome.
Solo se requiere una copia del gen mutado para que se manifieste su efecto. Curiosamente, en individuos varones XY, la presencia de una única copia del cromosoma X resulta ser letal antes del nacimiento. No obstante, los hombres XX (por ejemplo, cariotipos 47:XXY o 46:XX con una translocación Xp;Y) pueden sobrevivir y exhibir las características clínicas típicas del MLS.
Dado que las mujeres poseen dos copias del cromosoma X, las afectadas generalmente albergan una copia génica normal junto a una anormal o delecionada. Hasta el momento, la mayoría de las anomalías identificadas en casos de síndrome MLS involucran una deleción en una región específica del cromosoma X. Si bien se han reportado casos raros de mutaciones puntuales (sin sentido y sin-sentido), es importante notar que no todos los casos diagnosticados con MLS presentan una mutación confirmada en el material genético analizado.
Las alteraciones genéticas se han localizado en el gen HCCS, ubicado en Xp22.2, afectando a la holocitocromo-c sintetasa (también conocida como heme liasa). Esta enzima es fundamental dentro de las mitocondrias, participando en la producción de citocromo c, un componente vital para la fosforilación oxidativa y la inducción de la muerte celular programada (apoptosis).
Características Clínicas Distintivas del Síndrome MLS
Las manifestaciones clínicas principales del síndrome MLS se observan desde el nacimiento, afectando esencialmente a dos sistemas orgánicos clave: el ojo y la piel.
Manifestaciones Oculares: Microftalmia
La microftalmia es el término utilizado para describir ojos cuyo globo ocular es significativamente más pequeño de lo normal. Esta condición puede afectar a uno o a ambos ojos. Es importante señalar que no implica anoftalmia (la ausencia total de globos oculares), aunque en clasificaciones antiguas se usase dicho término incorrectamente en ciertos casos de MLS. Una característica ocular frecuente en el síndrome MLS es la opacidad de la córnea, conocida como esclerocórnea.
Defectos Cutáneos Lineales
Casi la totalidad de los pacientes presentan afectaciones cutáneas. Estas se caracterizan típicamente por defectos lineales irregulares, a menudo localizados en la región facial y cervical. Dichos patrones suelen seguir las Líneas de Blaschko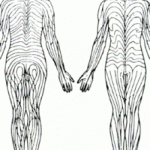 Comprendiendo las Líneas de Blaschko en Dermatología Las denominadas Líneas de Blaschko se conceptualizan como trayectorias específicas de migración y posterior proliferación celular epidérmica ocurridas durante el desarrollo fetal temprano. Historia y Descripción de las Líneas de Blaschko Este patrón fundamental fue introducido al ámbito científico por el dermatólogo alemán Alfred Blaschko, quien lo presentó formalmente en 1901 durante el VII Congreso de la Sociedad Dermatológica Alemana. Estas líneas definen más. Si bien estas líneas aparecen de coloración rojiza al nacer, evolucionan y sanan dejando zonas de pigmentación marrón permanente.
Comprendiendo las Líneas de Blaschko en Dermatología Las denominadas Líneas de Blaschko se conceptualizan como trayectorias específicas de migración y posterior proliferación celular epidérmica ocurridas durante el desarrollo fetal temprano. Historia y Descripción de las Líneas de Blaschko Este patrón fundamental fue introducido al ámbito científico por el dermatólogo alemán Alfred Blaschko, quien lo presentó formalmente en 1901 durante el VII Congreso de la Sociedad Dermatológica Alemana. Estas líneas definen más. Si bien estas líneas aparecen de coloración rojiza al nacer, evolucionan y sanan dejando zonas de pigmentación marrón permanente.
Otras Manifestaciones Asociadas al Síndrome MLS
Existen reportes de algunos pacientes que experimentaron únicamente signos oculares o solo anomalías cutáneas, presentándose a veces en combinación con otras características menos comunes.
El manejo integral del síndrome MLS requiere la evaluación detallada de todas sus posibles manifestaciones. Es crucial que el diagnóstico y seguimiento incluyan tanto la evaluación oftálmica especializada como la dermatológica para abordar las secuelas genéticas en estos sistemas primarios.
Otras características clínicas relevantes se detallan exhaustivamente en la siguiente tabla, resumiendo los hallazgos multisistémicos asociados con el síndrome MLS.
| Corazón |
|
| Cerebro |
|
| Ojo |
|
| Hernia de diafragma |
|
| Estatura |
|
| Sistema Urogenital |
|
Es importante notar que la manifestación clínica del síndrome MLS puede mostrar una variación considerable en su severidad y patrón, incluso entre miembros de la misma familia afectada.
Diagnóstico del Síndrome Microsílabas Labiales (MLS)
El diagnóstico inicial del síndrome MLS se establece fundamentalmente de manera clínica, basándose en la observación de las alteraciones distintivas presentes tanto en los ojos como en la piel del paciente.
Los estudios citogenéticos mediante cariotipo suelen revelar una disparidad notable en uno de los cromosomas X, caracterizándose típicamente por una deleción detectada en el brazo corto del cromosoma. Complementariamente, el gen HCCS puede ser analizado mediante secuenciación en laboratorios de investigación si la deleción cromosómica no resulta evidente en el análisis estándar.
En un caso reportado, la biopsia de una lesión cutánea reveló la presencia de un hamartoma de músculo liso.
Dada la presentación de anormalidades oculares y dérmicas, puede ser necesario diferenciar el síndrome MLS de la hipoplasia dérmica focal Comprendiendo la Hipoplasia Dérmica Focal Genética La hipoplasia dérmica focal (HDF) es un trastorno genético hereditario que fue identificado por primera vez por Goltz en 1962. Esta condición puede impactar el desarrollo de múltiples sistemas orgánicos, manifestándose típicamente con anormalidades en la piel, los ojos y los dientes. Además, su alcance puede extenderse al sistema esquelético, gastrointestinal, genitourinario, neurológico y cardiovascular. Se trata de una patología poco frecuente, con menos más o del síndrome de Goltz (MIM305600). Este último es otro síndrome ligado al cromosoma X que impacta principalmente a mujeres. No obstante, las anomalías esqueléticas como la sindactilia (fusión de dedos de manos o pies) o la hendidura de estas extremidades no se observan en el síndrome MLS. El gen responsable de la hipoplasia dérmica focal ha sido identificado en la región Xp11.23: el gen PORCN.
Comprendiendo la Hipoplasia Dérmica Focal Genética La hipoplasia dérmica focal (HDF) es un trastorno genético hereditario que fue identificado por primera vez por Goltz en 1962. Esta condición puede impactar el desarrollo de múltiples sistemas orgánicos, manifestándose típicamente con anormalidades en la piel, los ojos y los dientes. Además, su alcance puede extenderse al sistema esquelético, gastrointestinal, genitourinario, neurológico y cardiovascular. Se trata de una patología poco frecuente, con menos más o del síndrome de Goltz (MIM305600). Este último es otro síndrome ligado al cromosoma X que impacta principalmente a mujeres. No obstante, las anomalías esqueléticas como la sindactilia (fusión de dedos de manos o pies) o la hendidura de estas extremidades no se observan en el síndrome MLS. El gen responsable de la hipoplasia dérmica focal ha sido identificado en la región Xp11.23: el gen PORCN.
Adicionalmente, es crucial descartar el síndrome de Aicardi (MIM304050), ya que comparte afectaciones similares en ojo y cerebro, aunque este último se restringe exclusivamente a mujeres y no presenta afecciones cutáneas. Si bien el gen causante del síndrome de Aicardi aún no ha sido identificado, su localización se ha determinado en la banda Xp22.
Manejo y Tratamiento del Síndrome MLS
El tratamiento para el síndrome MLS es sintomático, enfocándose en abordar y manejar las complicaciones específicas a medida que van apareciendo. Actualmente, no existe una cura definitiva para esta condición.
Las lesiones cutáneas tienden a sanar espontáneamente, dejando como secuela pigmentación de color marrón permanente en la zona afectada.
Para obtener información detallada sobre la gestión y las últimas investigaciones relacionadas con el espectro del síndrome MLS, se recomienda consultar con especialistas genéticos y médicos enfocados en enfermedades raras.


