Entendiendo la Enfermedad de Wilson: Causas, Síntomas y Manifestaciones Orgánicas
La enfermedad de Wilson es un trastorno hereditario poco común caracterizado por la acumulación excesiva de cobre corporal. Este padecimiento afecta de manera desproporcionada, siendo aproximadamente cuatro veces más prevalente en mujeres que en hombres. Normalmente, el cobre se metaboliza eficazmente, integrándose en compuestos como las enzimas, concretamente la ceruloplasmina, y luego se excreta a través de la bilis.
Sin embargo, en la enfermedad de Wilson, este proceso metabólico se interrumpe. El exceso de cobre se deposita primariamente en el hígado, provocando daño a las células hepáticas. A medida que los niveles hepáticos de cobre aumentan progresivamente, este metal finalmente se libera al torrente sanguíneo y se acumula en otros órganos vitales, destacando el cerebro y la médula espinal. Estos depósitos tóxicos generan daño tisular, muerte celular y cicatrización, lo que compromete seriamente la función normal de los órganos afectados. Esta afección también es conocida históricamente como degeneración hepatolenticular.
Identificación de Signos y Síntomas de la Enfermedad de Wilson
Aunque la acumulación anormal de cobre comienza desde el nacimiento, los signos clínicos de la enfermedad de Wilson no suelen manifestarse hasta finales de la niñez o entrada la adolescencia. Dado que el hígado es el primer órgano receptor del exceso de cobre, las manifestaciones iniciales de la enfermedad típicamente están ligadas a este órgano e incluyen:
- Hepatitis (una inflamación hepática que puede ser aguda o crónica).
- Cirrosis (una enfermedad hepática severa resultante de la pérdida progresiva de la funcionalidad del hígado).
- Insuficiencia hepática fulminante (una falla hepática repentina, a menudo con desenlace mortal).
La presentación inicial más frecuente es la cirrosis, observándose generalmente durante los primeros diez años de vida. Los síntomas secundarios que pueden emerger a medida que progresa el daño hepático incluyen:
- Confusión y somnolencia debido a encefalopatía hepática.
- Hemorragia intestinal causada por la hipertensión portal debida a la compresión de venas dentro del hígado, llevando a varices esofágicas o gástricas, y consecuente hipertensión portal.
- Acumulación anormal de líquido en el abdomen (ascitis) e hinchazón en las piernas (edema).
- Aumento de la susceptibilidad a infecciones.
- Exceso de líquido pulmonar que dificulta la respiración.
Cuando el cobre comienza a acumularse en otros sistemas orgánicos, pueden manifestarse los siguientes conjuntos de síntomas neurológicos y sistémicos:
Manifestaciones Neurológicas (Generalmente en la Segunda o Tercera Década)
Los síntomas neurológicos suelen hacerse evidentes alrededor de la segunda o tercera década de vida, aunque pueden surgir incluso hasta los 50 años de edad:
- Temblores en cabeza, brazos o piernas.
- Deterioro del tono muscular causando movimientos lentos, impredecibles o repetitivos (distonía o corea).
- Alteraciones del habla (habla arrastrada o lenta), producción excesiva de saliva, y expresiones faciales lentificadas o disminuidas (lo que se conoce como facies en máscara).
- Cambios conductuales o emocionales significativos.
- Estados de confusión, delirio o desarrollo de demencia.
Síntomas Oculares Notables
Estos indicadores son cruciales para el diagnóstico temprano:
- Presencia de Anillos de Kayser-Fleischer: Son anillos visibles formados por depósitos de cobre en la membrana corneal. Su coloración puede variar entre dorado verdoso y tonos marrones.
Afectaciones Musculoesqueléticas
La rigidez y debilidad también son secuelas del depósito de cobre:
- Rigidez articular y dificultad para la movilidad de las extremidades a medida que las articulaciones se ven comprometidas.
- Debilitamiento y adelgazamiento progresivo de la estructura ósea.
Cambios Cutáneos
La piel y el aspecto externo también reflejan la toxicidad del cobre:
- Coloración amarillenta de la piel y los ojos (ictericia).
- Desarrollo de manchas cutáneas inusualmente oscuras (hiperpigmentación pigmentación).
- Otros signos asociados a la enfermedad hepática: telangiectasias de araña y eritema palmar (enrojecimiento
 Componentes y Diagnóstico del Enrojecimiento Facial El enrojecimiento, conocido médicamente como rubor, se produce cuando los vasos sanguíneos superficiales de la piel se dilatan. Si esta dilatación es mediada por la actividad de los nervios que inervan dichos vasos, frecuentemente se acompaña de sudoración. Por otro lado, los agentes que actúan directamente sobre la pared vascular provocan típicamente un rubor seco, sin transpiración. Imágenes Ilustrativas del Enrojecimiento Enrojecimiento inducido por más de las palmas de las manos).
Componentes y Diagnóstico del Enrojecimiento Facial El enrojecimiento, conocido médicamente como rubor, se produce cuando los vasos sanguíneos superficiales de la piel se dilatan. Si esta dilatación es mediada por la actividad de los nervios que inervan dichos vasos, frecuentemente se acompaña de sudoración. Por otro lado, los agentes que actúan directamente sobre la pared vascular provocan típicamente un rubor seco, sin transpiración. Imágenes Ilustrativas del Enrojecimiento Enrojecimiento inducido por más de las palmas de las manos).
Problemas Hematológicos Comunes
El cobre también impacta la producción y función de las células sanguíneas:
- Anemia hemolítica (destrucción acelerada de glóbulos rojos).
- Trombocitopenia (niveles bajos de plaquetas, lo cual aumenta el riesgo de sangrado).
La enfermedad de Wilson requiere diagnóstico y tratamiento tempranos para prevenir daños orgánicos irreversibles. Si se detecta a tiempo, la terapia quelante o los tratamientos basados en zinc pueden controlar eficazmente la sobrecarga de cobre y mejorar significativamente el pronóstico del paciente.
- (de plaquetas), lo que provoca hematomas y sangrado fácil debido a una coagulación deficiente.
Nefropatía:
- Los síntomas exhiben una gran variabilidad.
- Existe la posibilidad de desarrollar cálculos renales o vesicales.
- Puede manifestarse sangre en la orina (hematuria).
¿Cuál es la causa de la Enfermedad de Wilson? (Etiología Genética)
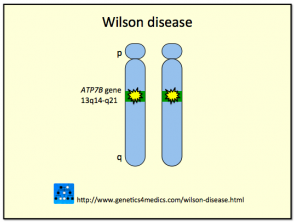
La Enfermedad de Wilson es desencadenada por mutaciones en el gen ATP7B, localizado en el brazo largo (q) del cromosoma 13. La proteína codificada por este gen —la adenosina trifosfatasa transportadora de cobre— es crucial para el transporte adecuado del cobre en el organismo. La herencia del gen defectuoso asociado a la Enfermedad de Wilson sigue un patrón autosómico recesivo. Esto implica que un individuo debe heredar una copia del gen mutado de ambos padres para desarrollar la afección. Aquellos que poseen un gen defectuoso y uno normal son clasificados como "portadores", permanecen completamente sanos y pueden transmitir el gen mutado a su descendencia.
Genética de la Enfermedad de Wilson *
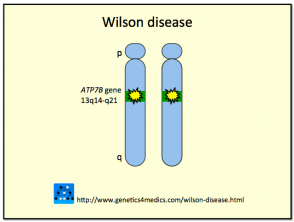
Enfermedad de Wilson
* Imagen cortesía de Genetics 4 Medics
Procedimientos de Diagnóstico para la Enfermedad de Wilson
Para establecer un diagnóstico definitivo de la Enfermedad de Wilson, se emplean diversas pruebas clínicas y de laboratorio. La presencia de anillos de Kayser-Fleischer, combinada con niveles de ceruloplasmina inferiores a 20 mg/dL en un paciente que presenta sintomatología neurológica, es fuertemente sugestiva de la enfermedad.
Los resultados analíticos comunes pueden incluir:
- Análisis de función hepática alterados: se observan niveles elevados de bilirrubina y de las enzimas transaminasas, junto con concentraciones bajas de albúmina circulante.
- Concentraciones bajas de ceruloplasmina sérica (aunque estas pueden ser normales en el 5-10% de los afectados por la Enfermedad de Wilson).
- Niveles de cobre sérico inferiores a 80 mcg/dL (a pesar de la acumulación tisular de cobre).
- Resultados de cobre urinario superiores a 100 mg.
- Niveles reducidos de ácido úrico en suero.
- Análisis histopatológico mediante biopsia hepática, que revela acumulación alta de cobre (más de 250 mcg por gramo de peso seco).
El examen físico puede evidenciar signos de afección hepática o esplénica, incluyendo cirrosis y necrosis del hígado. Las exploraciones radiológicas, como la Resonancia Magnética (RM) o la tomografía computarizada (CT) cerebral, pueden revelar anormalidades, particularmente en las estructuras de los ganglios basales del cerebro. Las exploraciones abdominales...
pueden indicar enfermedad hepática u otras anomalías.
Las pruebas genéticas para determinar la enfermedad de Wilson aún no están disponibles de forma rutinaria.
Directrices de Tratamiento para la Enfermedad de Wilson
El principal objetivo del tratamiento para la enfermedad de Wilson es doble: reducir la acumulación de cobre en los tejidos del cuerpo y gestionar eficazmente tanto los síntomas como las posibles complicaciones del trastorno.
Para lograr la eliminación del exceso de cobre corporal e inhibir la absorción continua de este mineral, se emplean agentes quelantes y medicamentos específicos que bloquean la absorción de cobre a nivel del tracto gastrointestinal.
- Acetato de zinc: Su función principal es impedir la absorción de cobre en el tracto gastrointestinal (GI).
- Penicilamina: Este agente forma complejos solubles con el cobre, facilitando su excreción a través de la orina.
- Trientina: Posee una acción similar a la penicilamina y constituye una alternativa viable para aquellos pacientes que no toleran la penicilamina. Este tratamiento debe administrarse concomitantemente con zinc.
Adicionalmente, se aconseja enfáticamente seguir una dieta con bajo contenido de cobre. Es crucial evitar el consumo de alimentos conocidos por su alta concentración de este mineral, tales como hígado, champiñones, chocolate, nueces, frutos secos y mariscos.
Los pacientes diagnosticados con enfermedad de Wilson invariablemente requerirán terapia continua y de por vida para gestionar este desorden crónico. Dado que la enfermedad de Wilson puede resultar fatal debido a la insuficiencia de la función hepática y los efectos tóxicos del cobre en el sistema nervioso, el monitoreo regular es esencial para abordar tempranamente cualquier complicación y prevenir secuelas graves.
En casos donde la enfermedad hepática progresa hacia una fase grave, el trasplante hepático puede ser ofrecido como una opción terapéutica definitiva.


