Entendiendo el Feocromocitoma: Definición, Prevalencia y Causas Genéticas
El feocromocitoma (a veces escrito feocromocitoma en la nomenclatura estadounidense) es un trastorno neuroendocrino poco común caracterizado por un tumor que produce y secreta cantidades excesivas de catecolaminas, principalmente noradrenalina y, en menor grado, adrenalina.
Estos tumores tienen su origen primario en la médula suprarrenal (aproximadamente el 85% de los casos). Los casos restantes (15%) se desarrollan a partir de neural ganglios ubicados en cabeza y cuello, y en estos escenarios se les denomina paragangliomas.
¿Quiénes son Susceptibles al Feocromocitoma?
El feocromocitoma es considerado una afección rara. Las estimaciones indican que se diagnostican entre 2 y 8 casos nuevos anualmente por cada millón de habitantes. Sin embargo, su prevalencia es mayor en poblaciones específicas; se encuentra en aproximadamente 1 a 6 de cada 1,000 pacientes que padecen hipertensión no explicada.
La media de edad al momento del diagnóstico se sitúa alrededor de los 40 años, aunque es crucial reconocer que esta condición puede manifestarse en cualquier etapa de la vida.
Factores Etiológicos del Feocromocitoma
La causa subyacente del feocromocitoma se clasifica de la siguiente manera:
- El 75% de los casos de feocromocitoma son esporádicos, es decir, no están vinculados a una condición familiar conocida.
- El 25% restante se atribuye a genético mutaciones hereditarias.
Los síndromes hereditarios y las mutaciones genéticas asociadas con una predisposición al feocromocitoma son significativos e incluyen los siguientes:
| Enfermedad Nombrada | Gen Afectado | Patrón de Herencia | Fenotipo Clínico Asociado |
|---|---|---|---|
| Enfermedad de Von Hippel-Lindau | Gen VHL en el cromosoma 3p25 | Autosómico dominante | Feocromocitomas (presentes en el 20% de los casos), angiomatosis, renal celda clara carcinomas, quistes renales, tumores neuroectodérmicos primitivos (TNEP), hemangioblastomas retinianos, tumores pancreáticos, tumores endolinfáticos, cistoadenomas epididimarios |
Neurofibromatosis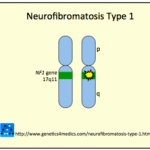 ¿Qué es la Neurofibromatosis? La neurofibromatosis (NF) es un trastorno genético que afecta al hueso, el tejido suave, la piel y el sistema nervioso. Las manifestaciones clínicas de esta condición suelen aumentar con el tiempo. Existen al menos 8 fenotipos clínicos diferentes de la NF. Generalmente, se clasifica en dos tipos principales: • Neurofibromatosis tipo 1 (NF1) • Neurofibromatosis tipo 2 (NF2). Neurofibromatosis Tipo 1 (NF1) La NF1 afecta aproximadamente más Tipo 1 ¿Qué es la Neurofibromatosis? La neurofibromatosis (NF) es un trastorno genético que afecta al hueso, el tejido suave, la piel y el sistema nervioso. Las manifestaciones clínicas de esta condición suelen aumentar con el tiempo. Existen al menos 8 fenotipos clínicos diferentes de la NF. Generalmente, se clasifica en dos tipos principales: • Neurofibromatosis tipo 1 (NF1) • Neurofibromatosis tipo 2 (NF2). Neurofibromatosis Tipo 1 (NF1) La NF1 afecta aproximadamente más Tipo 1 |
Gen NF1 en el cromosoma 17q11 | Dominante autosómico | Feocromocitoma (ocurre en el 1 a 3% de los casos), neurofibromas, dificultades de aprendizaje, escoliosis, cifosis |
| Múltiple Endocrino Neoplasia Tipo 2 (MEN 2) | Gen RET en el cromosoma 10q11 | Dominante autosómico | Feocromocitomas bilaterales (en 50-80% de los casos), carcinoma medular de tiroides, adenoma paratiroideo |
| Mutaciones en Genes del Complejo de la Proteína Succinato Deshidrogenasa Proteína Línea germinal | Gen SDHA en el cromosoma 5p15 | Dominante autosómico | Cardiomiopatía dilatada por paraganglioma, síndrome de Leigh |
| SDHAF2 en el cromosoma 11q12 | Dominante autosómico | Paraganglioma |
El diagnóstico preciso de estas condiciones genéticas es fundamental, ya que un porcentaje significativo de los feocromocitomas provienen de síndromes hereditarios que requieren vigilancia y manejo especializados.
| Gen | Ubicación Cromosómica y Tipo de Herencia | Síndromes Asociados |
|---|---|---|
| SDHB | Gen SDHB en el cromosoma 1p36, Autosómico Dominante | Feocromocitoma, paraganglioma, síndrome de Cowden, carcinomas de células renales, tumores del estroma gastrointestinal |
| SDHD | Gen SDHD en el cromosoma 11q23, Autosómico Dominante | Feocromocitoma, paraganglioma, tumores carcinoides, síndrome de Cowden, tumores del estroma gastrointestinal, sordera |
| TMEM127 | Gen TMEM127 en el cromosoma 2q11, Autosómico Dominante | Feocromocitomas bilaterales; la malignidad es poco frecuente |
| MAX | Gen MAX en el cromosoma 14q23, Autosómico Dominante | Feocromocitomas malignos |
Aproximadamente el 10% de las mutaciones genéticas identificadas se asocian con malignidad (cáncer).
Características Clínicas del Feocromocitoma y Paraganglioma
Los síntomas del feocromocitoma son notablemente variables, manifestándose típicamente en episodios paroxísticos debido a la liberación descontrolada de hormonas. Los signos clínicos más comunes incluyen:
- Cefaleas intensas
- Sudoración profusa (hiperhidrosis
 Comprendiendo la Hiperhidrosis: Síntomas, Causas y Población Afectada La hiperhidrosis se define como la sudoración excesiva e incontrolable. Este fenómeno va más allá de la transpiración normal necesaria para regular la temperatura corporal. El sudor es esencialmente una solución acuosa producida por las glándulas sudoríparas ecrinas. Estas glándulas se distribuyen por toda la superficie corporal, aunque se concentran significativamente en las palmas de las manos y las plantas de los más)
Comprendiendo la Hiperhidrosis: Síntomas, Causas y Población Afectada La hiperhidrosis se define como la sudoración excesiva e incontrolable. Este fenómeno va más allá de la transpiración normal necesaria para regular la temperatura corporal. El sudor es esencialmente una solución acuosa producida por las glándulas sudoríparas ecrinas. Estas glándulas se distribuyen por toda la superficie corporal, aunque se concentran significativamente en las palmas de las manos y las plantas de los más) - Palpitaciones y taquicardia
- Episodios de ansiedad o pánico
- Hipertensión paroxística (presión arterial elevada en picos)
- Casos severos de hipertensión maligna
La malignidad en estos tumores se diagnostica formalmente por la existencia de metástasis a órganos distantes. Las tasas de enfermedad metastásica oscilan entre el 10% y el 15% para el feocromocitoma, pero alcanzan el 20% al 50% en el caso de los paragangliomas. Los sitios primarios de diseminación metastásica incluyen el esqueleto, los pulmones, el hígado y los ganglios linfáticos.
Métodos de Diagnóstico para el Feocromocitoma
La sospecha clínica de feocromocitoma surge frecuentemente debido a la presentación sintomática clásica, especialmente en pacientes con hipertensión de origen desconocido. El diagnóstico se confirma mediante una serie de análisis bioquímicos y estudios de imagenología.
Análisis Bioquímicos
- Detección de catecolaminas y metanefrinas en plasma o mediante una recolección de orina de 24 horas: Niveles consistentemente elevados (entre 3 y 4 veces el rango normal) sugieren fuertemente el diagnóstico de feocromocitoma.
- Medición de Cromogranina A: Este marcador tumoral suele estar elevado en presencia de feocromocitoma.
- Prueba de supresión con clonidina: Utilizada para confirmar la etiología tumoral de la secreción de catecolaminas.
- Análisis genéticos: Se recomiendan cuando se detectan tumores múltiples o existe un historial familiar de feocromocitoma. Para tumores solitarios en pacientes mayores de 40 años sin antecedentes familiares, el estudio genético puede no ser estrictamente necesario.
Estudios de Imagenología para Localización Anatómica
La elección del método de imagen depende de la necesidad de evaluar la anatomía local o la funcionalidad del tumor.
- Resonancia Magnética (RM): Preferiblemente con contraste de gadolinio, la RM muestra típicamente una masa hiperintensa en la secuencia T2. Esta modalidad es superior a la TAC para evaluar la infiltración de estructuras vasculares y órganos adyacentes.
- Tomografía Computarizada (TC) con contraste: Sus ventajas radican en su menor costo, rápida disponibilidad y alta sensibilidad para detectar lesiones pequeñas (0.5 a 1 cm). No obstante, su especificidad para diferenciar el feocromocitoma de otros tejidos es limitada.
Las técnicas basadas en radionúclidos son fundamentales para determinar la actividad metabólica del tumor y son clave en el seguimiento post-tratamiento.
- Gammagrafía con metayodobencilguanidina marcada con yodo-123 (SPECT con I-123 MIBG): Posee una excelente especificidad y una sensibilidad alta (90-100%) para identificar tumores mayores de 1 a 2 cm. Es esencial para planificar la terapia dirigida con MIBG en casos de enfermedad diseminada.
- Tomografía por Emisión de Positrones (PET): Modalidades como PET-CT o PET-RM utilizando [18F]-L-dihidroxifenilalanina (18F-DOPA) ofrecen una sensibilidad y especificidad del 90-100% para tumores pequeños (> 1-2 cm). El 18F-DOPA proporciona una resolución espacial superior y una captación del radiotrazador más clara y selectiva en comparación con el 123I-MIBG. Su principal limitación es el mayor coste y menor disponibilidad.
Actualmente, los criterios para clasificar un tumor como maligno se basan en la producción hormonal excesiva y un tamaño superior a 4-6 cm. Es crucial notar que aún no existen criterios histológicos definitivos que permitan diferenciar con certeza entre tumores benignos y malignos.
Manejo y Tratamiento del Feocromocitoma
El tratamiento efectivo del feocromocitoma requiere la intervención de equipos multidisciplinarios, incluyendo endocrinólogos expertos, anestesistas y cirujanos. Este enfoque coordinado es fundamental para mitigar las complicaciones perioperatorias y reducir la morbilidad asociada al procedimiento.
Además, es crucial considerar la necesidad de asesoramiento genético para evaluar posibles síndromes hereditarios.
Control Farmacológico de la Hipertensión
Antes de la cirugía, el manejo riguroso de la hipertensión es esencial. Las estrategias médicas incluyen:
- Uso de alfabloqueantes, como la fenoxibenzamina (de acción prolongada) o la doxazosina (de acción corta).
- Administración de betabloqueantes, como el atenolol. Es vital nunca usarlos sin un bloqueo alfa concurrente, debido al alto riesgo de provocar una hipertensión refractaria.
Tratamiento del Feocromocitoma Localizado
La escisión quirúrgica constituye la única modalidad de tratamiento curativo para los tumores localizados. Optimizar el manejo de la presión arterial durante el periodo operatorio influye directamente en los resultados del paciente.
Generalmente, el abordaje laparoscópico es preferido sobre la cirugía abierta debido a sus ventajas. Es importante destacar que:
- La "técnica sin toque" retroperitoneal se considera superior al abordaje transperitoneal.
- Los beneficios de la laparoscopia sobre la cirugía abierta incluyen menor tiempo de hospitalización, reducción en la pérdida de sangre, una recuperación postoperatoria más rápida y mejores resultados estéticos.
- Existe un riesgo del 5% de requerir conversión a un enfoque abierto durante la laparoscopia.
- Las contraindicaciones para la laparoscopia incluyen tumores mayores a 8 cm, sospecha de malignidad u obesidad extrema (IMC superior a 45).
Abordaje del Feocromocitoma Metastásico
Cuando el feocromocitoma es metastásico, las opciones de tratamiento son limitadas y no curativas. Las estrategias se centran en el control de la enfermedad y la paliación de síntomas.
Las opciones de quimioterapia para el feocromocitoma metastásico incluyen:
- Régimen de ciclofosfamidaCiclofosfamida: Usos Dermatológicos y Consideraciones Cruciales del Tratamiento La ciclofosfamida es un agente farmacológico de gran potencia, utilizado primariamente en la terapia oncológica. Este medicamento pertenece a la clase de agentes citotóxicos conocidos como alquilantes, los cuales ejercen su acción al interferir directamente con la proliferación y replicación de las células malignas. Más allá de su aplicación en cáncer, la ciclofosfamida ha probado ser valiosa en el manejo de diversas más, vincristina y dacarbazina, con una supervivencia a cinco años reportada entre el 30 y el 60%.
- Uso de inhibidores del receptor de la tirosina quinasa, como el sunitinib.
- Fármacos inhibidores de mTOR, por ejemplo, everolimus.
- Tratamiento con 131I – MIBG.
Otras modalidades terapéuticas disponibles para el manejo del feocromocitoma metastásico comprenden:
- Radioterapia.
- Procedimientos de ablación.
- Cirugía para la reducción del volumen tumoral.
Pronóstico y Seguimiento Post-Tratamiento del Feocromocitoma
El pronóstico para los pacientes con feocromocitoma se ve negativamente afectado por ciertos factores, como:
- Estar cursando un embarazo.
- Presentar una edad avanzada.
No obstante, el pronóstico general es excelente cuando se logra la resección completa de un feocromocitoma esporádico, dado el bajo riesgo de recaída o malignidad.
Sin embargo, en las causas hereditarias, aproximadamente un tercio de los pacientes que presentan enfermedad extraadrenal experimentan una reaparición de la patología.
El plan de seguimiento posterior al tratamiento es riguroso y esencial para detectar recurrencias:
- Se aconseja un seguimiento clínico y analítico anual durante al menos 10 años tras la cirugía inicial.
- Para individuos con síntomas extraadrenales o con antecedentes de feocromocitoma familiar, se recomienda seguimiento de por vida.
- La presión arterial y los niveles de catecolaminas urinarias deben ser monitorizados regularmente.
- Las pruebas de imagen, como CT y/o MRI, deben realizarse aproximadamente cada dos años.


