Reglas Fundamentales: Comprendiendo el Síndrome de Peutz-Jeghers (PJS)
¿Qué es el Síndrome de Peutz-Jeghers?
El síndrome de Peutz-Jeghers (PJS) es una enfermedad hereditaria poco común. Se distingue por la presencia de pólipos gastrointestinales junto con una pigmentación característica que afecta tanto la piel como las membranas mucosas.
Los pólipos asociados al PJS son hamartomas; es decir, tumores benignos formados por una mezcla de células maduras propias del tejido donde se originan. Estos crecimientos pueden presentar cambios benignos (no cancerosos) o progresar a una naturaleza maligna (cancerosa). Los pacientes con PJS enfrentan un riesgo hasta 15 veces mayor de desarrollar alguna forma de cáncer interno en comparación con la población general.
¿Cuál es la causa genética del Síndrome de Peutz-Jeghers?
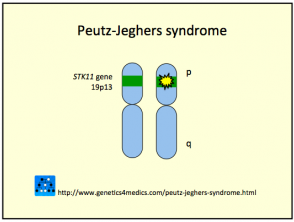
El síndrome de Peutz-Jeghers se origina por una línea germinal de mutaciones en el gen del supresor tumoral serina treonina quinasa, localizado en el cromosoma 19p13.3 (conocido también como STK11 / LKB1). Esta anomalía genética puede ser heredada o surgir de forma esporádica (en aproximadamente el 35% de los casos). Un factor crucial implicado es la reducción de la apoptosis, o la muerte celular programada, en las células afectadas.
El PJS sigue un patrón de herencia autosómica dominante. Esto implica que la descendencia de individuos afectados tiene una probabilidad del 50% de heredar la condición genética.
Análisis de la Genética del Síndrome de Peutz-Jeghers
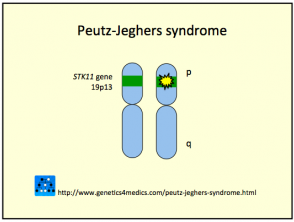
Síndrome de Peutz Jeghers
* Imagen cortesía de Genetics 4 Medics
Manifestaciones Clínicas del Síndrome de Peutz-Jeghers
Las manifestaciones clínicas asociadas al PJS se clasifican fundamentalmente en dos ámbitos: la afectación cutánea y las complicaciones gastrointestinales.
Hallazgos Cutáneos Distintivos
El rasgo cutáneo más reconocible del PJS es la aparición de máculas melanocíticas (manchas pigmentadas) en cerca del 95% de los pacientes. Estas se presentan como parches planos, cuyo color oscila entre canela, marrón oscuro y negro azulado, midiendo entre 1 y 5 mm. Su localización predilecta es alrededor de la boca, los labios, las encías, el revestimiento bucal interno, así como en manos, pies, dedos, y en la región del ano y genitales. Típicamente, esta pigmentación emerge antes de los 5 años y tiende a desaparecer gradualmente después de alcanzar la pubertad.
Complicaciones Gastrointestinales
Los pólipos gastrointestinales suelen manifestarse más tarde en la vida, siendo infrecuentes en la infancia. La presencia de estos pólipos incrementa el riesgo de complicaciones como hemorragias y dolor abdominal. Además, existe una alta probabilidad de que estos crecimientos evolucionen hacia un estado maligno.
La invaginación intestinal del intestino delgado (un proceso donde una porción del intestino se pliega sobre sí misma)
una porción de un intestino sobresale hacia otra) y la obstrucción intestinal también son complicaciones bastante comunes del síndrome de Peutz-Jeghers.
Métodos de Diagnóstico para el Síndrome de Peutz-Jeghers
El diagnóstico de esta afección se sospecha ante la presencia de las características clínicas típicas. Es importante diferenciarlo del síndrome de Laugier-Hunziker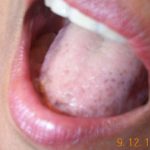 Entendiendo el Síndrome de Laugier-Hunziker: Características y Abordaje Definición y Diferenciación del Síndrome de Laugier-Hunziker El síndrome de Laugier-Hunziker es un trastorno esporádico y benigno que se manifiesta a través de marcas planas de pigmentación marrón en los labios y el interior de la boca. Frecuentemente, también se observan estrías marrones en las uñas. Es crucial diferenciar esta condición del síndrome de Peutz-Jeghers, ya que comparten características cutáneas similares, pero más, que se manifiesta con máculas pigmentadas similares alrededor y dentro de la boca, pero que frecuentemente también incluye melanoniquia
Entendiendo el Síndrome de Laugier-Hunziker: Características y Abordaje Definición y Diferenciación del Síndrome de Laugier-Hunziker El síndrome de Laugier-Hunziker es un trastorno esporádico y benigno que se manifiesta a través de marcas planas de pigmentación marrón en los labios y el interior de la boca. Frecuentemente, también se observan estrías marrones en las uñas. Es crucial diferenciar esta condición del síndrome de Peutz-Jeghers, ya que comparten características cutáneas similares, pero más, que se manifiesta con máculas pigmentadas similares alrededor y dentro de la boca, pero que frecuentemente también incluye melanoniquia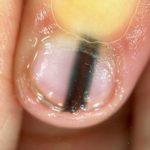 ```html Comprendiendo la Melanoniquia: Causas y Factores de Riesgo ¿Qué es la Melanoniquia? La melanoniquia se define como la aparición de una coloración marrón o negra en una uña. Esta pigmentación puede manifestarse de manera difusa o presentarse como una banda longitudinal a lo largo de la lámina ungueal. Ejemplos de Melanoniquia Longitudinal Benigna Revise más casos visuales de melanoniquia para una mejor comprensión del diagnóstico. ¿Quiénes corren el riesgo más (bandas pigmentadas longitudinales en una o más uñas). El síndrome de Laugier-Hunziker no está asociado a mutaciones del gen STK11.
```html Comprendiendo la Melanoniquia: Causas y Factores de Riesgo ¿Qué es la Melanoniquia? La melanoniquia se define como la aparición de una coloración marrón o negra en una uña. Esta pigmentación puede manifestarse de manera difusa o presentarse como una banda longitudinal a lo largo de la lámina ungueal. Ejemplos de Melanoniquia Longitudinal Benigna Revise más casos visuales de melanoniquia para una mejor comprensión del diagnóstico. ¿Quiénes corren el riesgo más (bandas pigmentadas longitudinales en una o más uñas). El síndrome de Laugier-Hunziker no está asociado a mutaciones del gen STK11.
El protocolo diagnóstico debe abarcar:
- Evaluación clínica de las lesiones pigmentarias.
- Recuento completo de células sanguíneas (CBC), dado que los pólipos pueden causar sangrado y derivar en anemia.
- Endoscopia exploratoria para identificar la existencia y localización de los pólipos.
- Examen patológico de los pólipos para confirmar el diagnóstico.
- Prueba genética del gen STK11, si está disponible en el centro médico.
Opciones de Tratamiento para el Síndrome de Peutz-Jeghers
Aproximadamente el 50% de los pacientes con PJS desarrollan y fallecen a causa de cáncer antes de los 57 años. El riesgo general de que los individuos con el síndrome de Peutz-Jeghers desarrollen algún tipo de cáncer durante su vida adulta asciende al 93%. Estos tumores no se limitan estrictamente al tracto gastrointestinal, sino que pueden surgir en múltiples localizaciones corporales, incluyendo mama, ovario, testículo, páncreas, útero, esófago y pulmón.
Actualmente, no existe un tratamiento específico curativo para el SPJ. El objetivo principal del manejo se centra en controlar y prevenir las complicaciones asociadas, como la obstrucción y la invaginación intestinal, así como el desarrollo oncológico.
El plan de manejo para pacientes con PJS debe incorporar:
- CBC anual de control.
- Examen físico anual enfocado en mamas, abdomen, pelvis y testículos.
- Extirpación frecuente de pólipos que causen sangrado o que sean grandes (> 5 mm) mediante polipectomía endoscópica.
- Laparotomía y resección quirúrgica cuando sean necesarias debido a invaginación intestinal persistente, obstrucción o sangrado recurrente.
- Extirpación quirúrgica de los cánceres tan pronto como se diagnostiquen.
Se puede ofrecer a los miembros familiares una prueba de detección de ADN para determinar si han heredado la mutación genética. Si es positivo, ellos también deben someterse a programas de detección periódica de enfermedades.
Las pecas cutáneas pueden volverse menos notorias mediante una protección solar rigurosa. En ciertos casos, la pigmentación puede atenuarse con tratamientos cosméticos. Además, el camuflaje estético también puede proporcionar un beneficio visual.


