(Histiocitosis sinusal con linfadenopatía masiva)
Introducción
La enfermedad de Rosai-Dorfman (ERD) es una entidad poco frecuente dentro del grupo de las histiocitosis no Langerhans. Aunque la forma clásica se caracteriza por una linfadenopatía masiva, especialmente cervical, la piel constituye el sitio extranodal más habitual de presentación. En dermatología, la variante cutánea aislada merece especial atención debido a su comportamiento clínico distintivo, su variabilidad morfológica y los desafíos diagnósticos que plantea. Conocer sus características es fundamental para diferenciarla de otras histiocitosis y de procesos infiltrativos o linfoproliferativos.
Definición y características clínicas
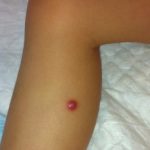
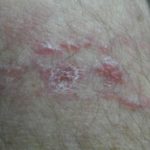
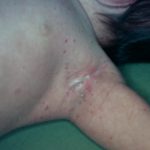
La ERD cutánea corresponde a una proliferación no maligna de histiocitos que infiltran la dermis y/o el tejido subcutáneo. Suele afectar a mujeres de edad media o avanzada y puede presentarse con:
- Pápulas solitarias
- Nódulos múltiples
- Placas infiltradas
Las lesiones varían en color (amarillento, eritematoso o marronáceo) y tamaño. A diferencia de la forma sistémica, la afectación cutánea aislada no suele acompañarse de linfadenopatías prominentes y tiende a cursar de manera más indolente.
Fisiopatología
La fisiopatología exacta de la ERD permanece incompletamente comprendida. Se considera un trastorno proliferativo reactivo de histiocitos derivados de la médula ósea, posiblemente desencadenado por estímulos inmunológicos o infecciosos. El rasgo citohistológico más característico, la emperipolesis, sugiere una alteración funcional de los histiocitos, que capturan células inflamatorias sin destruirlas. Algunos estudios han propuesto un vínculo con enfermedades relacionadas con IgG4, especialmente en casos con fibrosis marcada y abundantes células plasmáticas.
Epidemiología y factores de riesgo
- Es una enfermedad rara.
- La variante cutánea representa un porcentaje menor de los casos globales.
- Se observa predominantemente en mujeres mayores.
- No existen factores de riesgo claramente establecidos, aunque se han descrito asociaciones esporádicas con trastornos autoinmunes y procesos inflamatorios crónicos.
Diagnóstico clínico
El diagnóstico es principalmente histopatológico, pero el reconocimiento clínico-orientativo es clave.
Hallazgos clínicos típicos
- Lesiones firmes, bien delimitadas.
- Distribución variable, a menudo en tronco o extremidades.
- Ausencia de síntomas sistémicos en la forma cutánea pura.
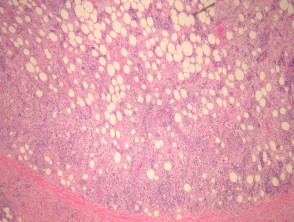
Hallazgos histológicos
El examen microscópico revela:
- Infiltración de la dermis y/o tejido subcutáneo con esclerosis variable.
- Histiocitos grandes de tipo epitelioide, con citoplasma pálido a eosinofílico.
- Presencia ocasional de formas multinucleadas.
- Mezcla con linfocitos, células plasmáticas, neutrófilos y eosinófilos.
Emperipolesis (signo distintivo)
La emperipolesis consiste en la presencia de linfocitos y otras células inflamatorias íntegras dentro del citoplasma de los histiocitos. Este hallazgo es fundamental para el diagnóstico diferencial, aunque no está presente en todos los campos ni en todas las muestras.
Células plasmáticas e IgG4
En la variante cutánea, las células plasmáticas pueden ser especialmente abundantes. Esto ha motivado la hipótesis de una posible superposición parcial con enfermedades del espectro de IgG4, aunque esta relación sigue siendo objeto de estudio.
Inmunohistoquímica
Los histiocitos característicos muestran:
- CD68: positivo
- S-100: positivo
- CD1a: negativo (útil para descartar histiocitosis de células de Langerhans)
Diagnóstico diferencial
El diagnóstico diferencial incluye otras histiocitosis y procesos linfoproliferativos:
1. Histiocitosis de células de Langerhans
- Positividad para CD1a y Langerina (CD207).
- Morfología distinta y ausencia de emperipolesis.
2. Reticulohistiocitoma
- Citoplasma típico en “vidrio esmerilado”.
- Generalmente solitario; no presenta emperipolesis.
3. Xantogranuloma juvenil
- Presencia de células gigantes de Touton.
- Marcada lipidización de los histiocitos.
4. Histiocitosis menos comunes
Pueden requerir panel inmunohistoquímico más amplio y correlación clínica rigurosa.
5. Linfoma T subcutáneo tipo paniculitis
- Infiltrado predominantemente linfocítico.
- Histiocitos con detritos nucleares fagocitados (“bean bag cells”), que pueden confundirse superficialmente con emperipolesis.
- Linfocitos con atipia nuclear y disposición típica bordeando adipocitos.
Tratamiento
La ERD cutánea suele tener un curso benigno y puede remitir espontáneamente. Sin embargo, el tratamiento se individualiza según extensión, síntomas y compromiso estético.
Opciones terapéuticas
- Corticosteroides tópicos o intralesionales
- Útiles en lesiones pequeñas o inflamadas.
- Corticosteroides sistémicos
- Indicación limitada a casos extensos o refractarios.
- Cirugía
- Efectiva para lesiones únicas o bien delimitadas.
- Inmunomoduladores
- Talidomida, dapsona o metotrexato se han utilizado en casos seleccionados.
- Radioterapia
- Considerada excepcionalmente en lesiones sintomáticas no resecables.
La evidencia es limitada debido a la rareza de la enfermedad; la elección depende del juicio clínico y la respuesta individual.
Manejo, pronóstico y seguimiento
- La variante cutánea aislada tiene pronóstico excelente.
- Puede persistir durante meses o años, con curso crónico pero benigno.
- Las recidivas son posibles, especialmente en lesiones múltiples.
- El seguimiento dermatológico periódico permite evaluar progresión, respuesta al tratamiento y descartar manifestaciones sistémicas.
Prevención y educación para el paciente
No existe prevención específica, ya que la etiología es desconocida. Sin embargo, es útil:
- Explicar la naturaleza benigna de la enfermedad.
- Aclarar que la variante cutánea no suele evolucionar a compromiso sistémico.
- Indicar la importancia de controles dermatológicos regulares.
Conclusión
La enfermedad de Rosai-Dorfman cutánea es una histiocitosis no Langerhans rara pero importante en la práctica dermatológica. Su diagnóstico se fundamenta en la correlación clínico-histológica, especialmente en la identificación de la emperipolesis y el perfil inmunohistoquímico característico. Aunque su curso suele ser benigno, un enfoque estructurado que incluya un adecuado diagnóstico diferencial y un plan terapéutico individualizado asegura un manejo óptimo y claro para pacientes y profesionales de la salud.
Patología de la enfermedad de Rosai-Dorfman
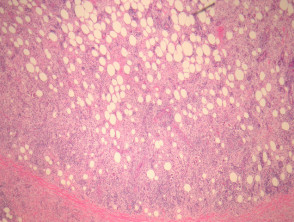
Figura 1
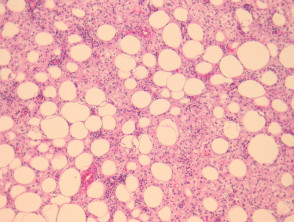
Figura 2
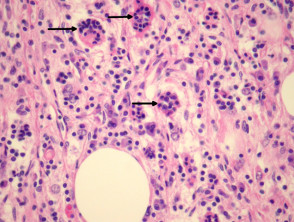
figura 3
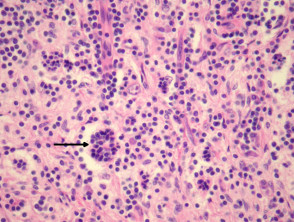
Figura 4
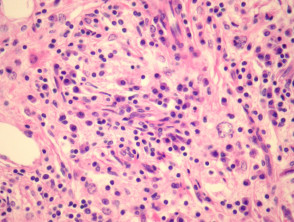
Figura 5