¿Qué es Peutz-Jeghers? síndrome?
El síndrome de Peutz-Jeghers (PJS) es una enfermedad hereditaria rara que se caracteriza por pólipos gastrointestinales en asociación con pigmentación afectando la piel y mucoso membranas.
Los pólipos PJS son hamartomas es decir benigno tumores compuestos por una mezcla de células maduras que normalmente se encuentran en ese tejido. Pueden mostrar benignos (no cancerosos) o maligno cambios (cancerosos). El riesgo de desarrollando alguna forma de interna cáncer es 15 veces mayor para los pacientes con PJS en comparación con la población general.
¿Cual es la causa?
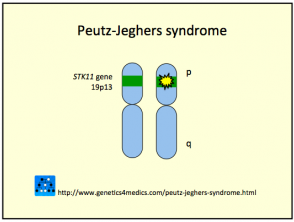
El síndrome de Peutz-Jeghers se debe a la línea germinal mutaciones en el supresor tumoral de serina treonina quinasa gene encontrado en cromosoma 19p13.3 (también llamado STK11 / LKB1). El gen anomalía pueden heredarse o surgir esporádicamente (35%). El factor clave puede reducirse apoptosis, o muerte celular programada, en las células afectadas.
PJS tiene autosómico herencia dominante. Esto significa que la descendencia de las personas afectadas tiene un 50% de posibilidades de heredar la enfermedad.
Genética del síndrome de Peutz-Jeghers *
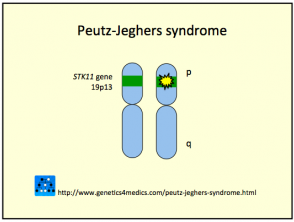
Síndrome de Peutz Jeghers
* Imagen cortesía de Genetics 4 Medics
¿Cuáles son las características clínicas del síndrome de Peutz-Jeghers?
Las características clínicas de PJS se pueden dividir en dos tipos, cutáneo y gastrointestinal.
Rasgos cutáneos
La característica cutánea más notable de PJS es la aparición de melanocítico máculas (pigmentado manchas) en el 95% de los pacientes. Se ven parches planos de color canela, marrón oscuro o negro azulado de 1 a 5 mm de tamaño alrededor de la boca, labios, encías, revestimiento interno de la boca, ojos, manos y pies, dedos de manos y pies. ano y áreas genitales. La pigmentación suele aparecer antes de los 5 años de edad y puede desaparecer después de la pubertad.
Características gastrointestinales
Los pólipos gastrointestinales aparecen más adelante en la vida y son raros en la niñez. Los pólipos pueden causar sangrado y dolor abdominal. Tienen una alta probabilidad de volverse malignos.
La invaginación intestinal del intestino delgado (cuando una porción de un intestino sobresale hacia otra) y la obstrucción intestinal también son complicaciones bastante comunes del síndrome de Peutz-Jeghers.
¿Cómo se diagnostica el síndrome de Peutz-Jeghers?
El diagnóstico se sospecha cuando existen características clínicas típicas. El síndrome de Laugier-Hunziker se presenta con máculas pigmentadas similares alrededor y dentro de la boca, pero a menudo también se presenta con melanoniquia (longitudinal bandas pigmentadas en uno o más uñas). El síndrome de Laugier-Hunziker no está asociado con STK11 mutaciones genéticas.
Diagnóstico trabajo Debería incluir:
- Evaluación clínica de pigmentario lesiones
- Recuento completo de células sanguíneas (CBC), ya que los pólipos pueden sangrar y provocar anemia
- Exploratorio endoscopia para determinar la presencia y ubicación de pólipos
- Patológico examen de pólipos para confirmar el diagnóstico
- STK11 prueba genética, si está disponible
¿Cuál es el tratamiento para el síndrome de Peutz-Jeghers?
Aproximadamente el 50% de los pacientes con PJS desarrollar y muere de cáncer a los 57 años. El riesgo general de que los pacientes con síndrome de Peutz-Jeghers desarrollen un cáncer durante la vida adulta es del 93%. Los cánceres no solo se encuentran en el tracto gastrointestinal, sino que pueden ocurrir en muchos otros sitios, incluidos los de mama, ovario, testículo, páncreas, útero, esófago y pulmón.
No existe un tratamiento específico para el SPJ, pero el objetivo principal es controlar y prevenir los problemas asociados de obstrucción intestinal e invaginación intestinal y cáncer. desarrollo.
El manejo de los pacientes con PJS debe incluir:
- CBC anual
- Examen físico anual de mamas, abdomen, pelvis y testículos
- Extracción repetida de sangrado o pólipos grandes (> 5 mm) mediante endoscópico polipectomía
- Laparotomía y resección según sea necesario para repetidos o persistente intususcepción intestinal, obstrucción o sangrado persistente
- Extirpación quirúrgica de cánceres a medida que se diagnostican
ADN Se puede ofrecer una prueba de detección a los miembros de la familia para ver si han heredado el gen. mutación. Si es así, también deben someterse a exámenes periódicos de detección de enfermedades.
Las pecas pueden ser menos obvias con una protección solar cuidadosa. En algunos casos, la pigmentación puede disminuir con un tratamiento cosmético. El camuflaje cosmético también puede ser útil.