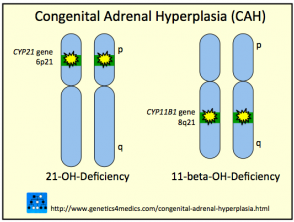
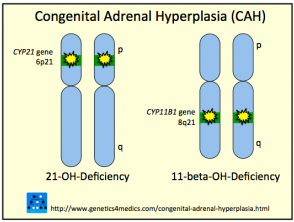
Enzimas Clave Implicadas en la Hiperplasia Suprarrenal Congénita
La Hiperplasia Suprarrenal Congénita (HSC) es un grupo de trastornos genéticos que afectan la producción de esteroides en las glándulas suprarrenales. Estas condiciones son el resultado de deficiencias enzimáticas hereditarias esenciales para la síntesis de cortisol y/o aldosterona. Las principales deficiencias enzimáticas responsables de la HSC incluyen:
- Deficiencia severa de la enzima 21-hidroxilasa.
- Déficit de la enzima 17-hidroxilasa.
- Insuficiencia de la 3β-hidroxiesteroide deshidrogenasa.
- Varias deficiencias enzimáticas parciales y raras.
Las manifestaciones clínicas observadas en los pacientes con HSC son altamente variables y dependen fundamentalmente de la enzima específica ausente o deficiente, así como del grado de actividad residual enzimática que presente el individuo.
Deficiencia de 21-Hidroxilasa: La Forma Más Prevalente de HSC
La 21-hidroxilasa es la enzima más comúnmente afectada, siendo responsable de aproximadamente el 95% de todos los diagnósticos de hiperplasia suprarrenal congénita. Esta deficiencia puede manifestarse desde el periodo neonatal temprano o permanecer latente hasta la pubertad o etapas adultas. La insuficiencia de esta enzima provoca una caída en la producción de cortisol y, simultáneamente, un incremento en la producción de andrógenos debido al desvío de precursores. Además, cerca de un tercio de los afectados sufren una síntesis inadecuada de aldosterona.
Cuando la forma grave de la deficiencia de 21-hidroxilasa se establece en las primeras dos semanas de vida, puede desencadenar una crisis de «pérdida de sal» —esencialmente, una insuficiencia suprarrenal aguda severa—. En el caso de las mujeres nacidas con la condición, se observa ambigüedad genital al nacer. Las variantes enzimáticas leves o parciales tienen un curso clínico más moderado, manifestándose típicamente más tarde con signos claros de exceso androgénico.
| Presentación Clínica Asociada a la Deficiencia de 21-Hidroxilasa |
| Crisis de Insuficiencia Suprarrenal Aguda |
- Resultado directo de la baja producción de cortisol junto con la deficiencia de aldosterona.
- Síntomas comunes: vómitos persistentes, deshidratación significativa y marcada hipotensión.
- Se presenta con un intenso deseo de ingerir sal (polidipsia salina).
|
| Ambigüedad Genital en el Recién Nacido |
- Provocada por la acumulación excesiva de andrógenos prenatales.
- Afecta únicamente a las pacientes de sexo femenino (causando pseudohermafroditismo femenino).
- Presentación típica: agrandamiento significativo del clítoris, con potencial fusión parcial o total de los labios mayores.
- Los genitales externos pueden asemejarse a estructuras típicamente masculinas.
|
| Manifestaciones de Inicio Tardío en Mujeres |
- Virilización progresiva: voz más grave, desarrollo mamario limitado, aumento de la masa muscular y libido elevada.
- Maduración esquelética acelerada (aceleración de la edad ósea).
- Irregularidades menstruales o amenorrea (falta de períodos).
- Incremento en el crecimiento de vello corporal según patrón masculino (hirsutismo
 Entendiendo el Hirsutismo: Definición y Causas El hirsutismo se define como un patrón de crecimiento piloso de tipo masculino que se manifiesta en mujeres después de la pubertad. Este crecimiento se localiza típicamente en áreas como el bigote y la barbilla, zonas donde el vello terminal aparece en hombres durante la pubertad junto con el vello axilar y púbico. Las mujeres que padecen esta condición también pueden experimentar un desarrollo más). Entendiendo el Hirsutismo: Definición y Causas El hirsutismo se define como un patrón de crecimiento piloso de tipo masculino que se manifiesta en mujeres después de la pubertad. Este crecimiento se localiza típicamente en áreas como el bigote y la barbilla, zonas donde el vello terminal aparece en hombres durante la pubertad junto con el vello axilar y púbico. Las mujeres que padecen esta condición también pueden experimentar un desarrollo más).
- Piel seborreica y acné
 Dominando el Conocimiento: ¿Qué es el Acné y Cómo se Manifiesta? El acné es un trastorno crónico que afecta primariamente al folículo piloso y a la glándula sebácea. Esta condición se caracteriza por la dilatación y obstrucción de estos folículos, acompañada frecuentemente de inflamación. Existen diversas manifestaciones y variantes de esta afección cutánea. ¿Quiénes son Susceptibles de Padecer Acné? El acné tiene un alcance universal, afectando a hombres y mujeres más resistente. Dominando el Conocimiento: ¿Qué es el Acné y Cómo se Manifiesta? El acné es un trastorno crónico que afecta primariamente al folículo piloso y a la glándula sebácea. Esta condición se caracteriza por la dilatación y obstrucción de estos folículos, acompañada frecuentemente de inflamación. Existen diversas manifestaciones y variantes de esta afección cutánea. ¿Quiénes son Susceptibles de Padecer Acné? El acné tiene un alcance universal, afectando a hombres y mujeres más resistente.
- Posible desarrollo de alopecia androgenética.
|
| Manifestaciones de Inicio Tardío en Hombres |
- Desarrollo puberal precoz (pubertad precoz).
- Aumento de la velocidad de maduración ósea.
- Agrandamiento testicular debido al crecimiento de tejido suprarrenal heterotópico dentro del parénquima testicular.
|
Proceso Diagnóstico de la Deficiencia de 21-Hidroxilasa
El diagnóstico preciso de la deficiencia enzimática requiere un enfoque metódico, comenzando por la evaluación clínica de los síntomas hormonales. La confirmación definitiva se basa en pruebas bioquímicas específicas.
Diagnóstico y Manejo de la Hiperplasia Suprarrenal Congénita (HSC)
El diagnóstico de la Hiperplasia Suprarrenal Congénita (HSC) debe considerarse en cualquier lactante que presente indicios de insuficiencia suprarrenal grave, virilización (en el caso de recién nacidas), o pubertad precoz (en varones).
Los análisis iniciales, solicitados ante una sospecha de insuficiencia suprarrenal aguda, revelarán el estado de pérdida de sal, el cual se caracteriza por los siguientes desequilibrios electrolíticos:
- Sodio sérico bajo (hiponatremia).
- Potasio sérico elevado (hiperpotasemia).
- Niveles bajos de aldosterona sérica.
- Concentraciones disminuidas de cortisol sérico.
- Actividad elevada de renina plasmática.
Para confirmar específicamente la deficiencia de 21-hidroxilasa, la enzima comúnmente afectada, se requiere medir la elevación de las siguientes hormonas en muestras de sangre u orina:
- 17-hidroxiprogesterona en suero.
- Sulfato de DHEA en plasma.
- Pregnanetriol en orina.
- 17-cetoesteroides medidos en orina.
Si los genitales de un recién nacido presentan anomalías morfológicas, puede ser necesaria una exploración mediante ultrasonido suprarrenal. Adicionalmente, las pruebas genéticas avanzadas están disponibles para identificar con precisión la mutación específica responsable de la condición.
Evaluación Diagnóstica Prenatal
Cuando se conoce que existe riesgo fetal debido a que un hermano anterior ha sido diagnosticado con HSC, se pueden contemplar pruebas genéticas durante la gestación para evaluar la salud del feto en desarrollo.
Asegurar la identificación temprana de la enzima deficiente es fundamental para implementar un manejo adecuado de la hiperplasia suprarrenal congénita, dado que el tratamiento varía sustancialmente según la causa subyacente y la gravedad de la disfunción hormonal.
Este escenario diagnóstico se confirma si ambos progenitores portan la anomalía genética. Las herramientas diagnósticas prenatales disponibles incluyen:
- Muestreo de vellosidades coriónicas, siendo el procedimiento recomendado a partir de la octava semana de gestación.
- Amniocentesis, que se realiza habitualmente cerca de la duodécima semana gestacional.
Asimismo, el diagnóstico definitivo puede obtenerse si se detectan concentraciones elevadas de 17-hidroxiprogesterona en el líquido amniótico, usualmente alrededor de la semana 14 de gestación.
Tratamiento de la Hiperplasia Suprarrenal Congénita Clásica
El manejo terapéutico de la forma clásica de HSC está enfocado en contrarrestar la secreción exagerada de ACTH mediante la administración de reposición de glucocorticoide (cortisol). Esto se logra habitualmente con una dosis baja de dexametasona, específicamente 0.5 mg administrados por la noche.
Para reducir eficazmente los niveles de andrógenos circulantes excesivos, se puede incorporar tratamiento antiandrogénico. Los agentes farmacológicos empleados comúnmente en estos casos incluyen:
- Acetato de ciproterona
- Espironolactona
- Flutamida
- FinasteridaEl Rol de la Testosterona y Andrógenos en la Caída del Cabello de Patrón Masculino La pérdida de cabello de patrón masculino, clínicamente conocida como alopecia androgenética, depende fundamentalmente de factores hormonales. En individuos con predisposición genética, la dihidrotestosterona (DHT), un potente metabolito derivado de la testosterona (la principal hormona testosterona/ andrógenos), es el principal culpable del adelgazamiento y eventual caída del cabello. Esta conversión crucial de testosterona a DHT más
En el subtipo específico que implica pérdida de sal, es un pilar del tratamiento fundamental administrar un mineralocorticoide, normalmente fludrocortisona (a una dosis estándar de 0.1 mg). El objetivo es mantener el volumen adecuado de líquido extracelular y estabilizar los niveles electrolíticos. La adherencia y eficacia del tratamiento se supervisan periódicamente mediante el control de la presión arterial, los electrolitos séricos y la actividad de la renina plasmática.
Identificación y Terapia para la Deficiencia de 17-Hidroxilasa
La deficiencia de 17-hidroxilasa representa una entidad patológica infrecuente que tiende a manifestarse durante la etapa puberal, debido a la producción reducida de andrógenos suprarrenales, lo que provoca hipogonadismo.
- En individuos con un fenotipo femenino, se observa un retraso significativo en la pubertad, manifestado por amenorrea primaria y ausencia de desarrollo mamario o púbico.
- En personas con un fenotipo masculino, se presentan genitales externos ambiguos o un aspecto externo predominantemente femenino (lo que se conoce como pseudohermafroditismo masculino).
Esta deficiencia enzimática resulta en una síntesis disminuida de cortisol y, concurrentemente, en un aumento de los niveles de mineralocorticoides. Los hallazgos analíticos distintivos incluyen:
- Presión arterial elevada (hipertensión).
- Concentraciones séricas bajas de potasio (hipopotasemia).
- Niveles reducidos de renina plasmática.
- Bajos niveles de 17 cetoesteroides excretados en la orina.
- Concentraciones elevadas de gonadotropina urinaria.
El enfoque terapéutico primario consiste en la administración de dexametasona para controlar la hipertensión asociada y la provisión de testosterona exógena para facilitar el desarrollo sexual apropiado y la maduración.
Comprensión Integral de la Deficiencia de 3-$beta$-Hidroxiesteroide Deshidrogenasa
La deficiencia de la enzima 3$beta$-hidroxiesteroide deshidrogenasa representa un trastorno endocrino significativo que obstruye la producción de todas las hormonas esteroides vitales generadas en las glándulas suprarrenales, incluyendo el cortisol, la aldosterona, los andrógenos y los estrógenos.
Esta condición patológica suele manifestarse de manera aguda durante la primera infancia, presentando síntomas críticos como episodios recurrentes de vómitos, pérdida severa de sales (crisis de pérdida de sal) y, en el caso de recién nacidos con predisposición genética, ambigüedad genital.
Los análisis bioquímicos son cruciales para el diagnóstico y típicamente revelan un patrón distintivo:
- Niveles bajos de sodio sérico.
- Concentraciones elevadas de potasio (hiperpotasemia).
- Un marcado incremento en la excreción urinaria de DHEA.
- Disminución de los metabolitos urinarios del cortisol, específicamente los 17 hidroxicorticosteroides.
Mecanismo Fisiopatológico del Bloqueo Enzimático
El núcleo del problema radica en la alteración de la conversión enzimática esencial de pregnenolona a progesterona. Este bloqueo metabólico es el responsable de coartar la síntesis adecuada tanto de cortisol como de aldosterona. Como consecuencia directa de esta insuficiencia, se produce una acumulación relativa y desequilibrada de andrógenos suprarrenales de menor potencia.
En el sexo femenino, este exceso de andrógenos resulta en grados variables de virilización parcial, afectando el desarrollo sexual secundario. Para los varones, la deficiencia en la producción testicular de testosterona causada por este defecto enzimático puede conducir a una formación incompleta o atípica de los genitales masculinos externos.
Estrategias Terapéuticas para la Insuficiencia Esteroidal
El manejo de la deficiencia de 3$beta$-hidroxiesteroide deshidrogenasa exige un plan terapéutico integral y de por vida. Este tratamiento debe incluir terapia de sustitución con glucocorticoides para compensar la falta de cortisol y suplementos de fludrocortisona para mantener el equilibrio electrolítico mediante la sustitución de aldosterona.
Además, es imperativo administrar esteroides sexuales apropiados para el sexo genético del individuo, comenzando su administración tan pronto como se alcance la etapa puberal para asegurar un desarrollo físico y sexual secundario acorde a las expectativas.