Cos'è la malattia di Fabry?
La malattia di Fabry è una malattia ereditaria lisosomiale disturbo da accumulo [1]. Conosciuto anche come malattia di Anderson-Fabry e angiocheratoma corpos diffuso.
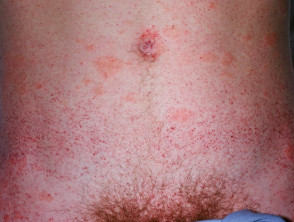
La malattia di Fabry provoca grappoli di angiocheratomi (piccole macchie rosso scuro sulla pelle) e molti sistemico sintomi dovuti a dichiarazione di globotriaosilceramide (Gb3) in più organi.
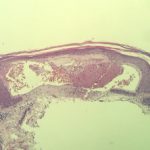
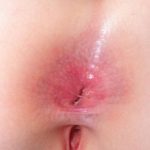
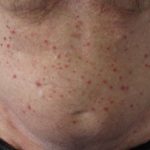
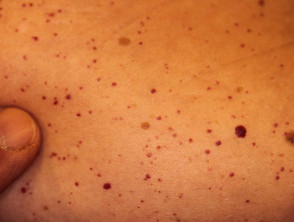
Angiocheratomi per sospetta malattia di Fabry
Angiocheratomi per sospetta malattia di Fabry
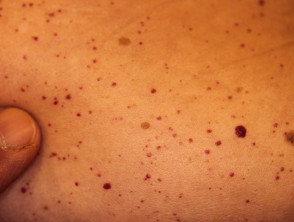
Angiocheratomi per sospetta malattia di Fabry
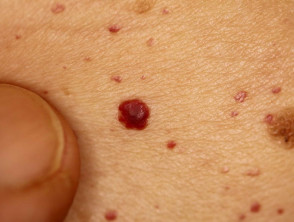
Angiocheratomi nella malattia di Fabry
Chi prende la malattia di Fabry?
Il preventivo predominanza negli uomini è circa uno su 40.000 a 60.000 della popolazione [2]. La malattia di Fabry colpisce generalmente uomini e ragazzi in modo più grave e in età più giovane rispetto alle donne e alle ragazze perché la sua eredità è legata all'X (maschio sesso ha solo una X cromosoma mentre le femmine ne hanno due). La malattia di Fabry può verificarsi in tutti i gruppi etnici. [3].
L'età di insorgenza della malattia di Fabry può variare dalla seconda alla quinta decade di vita o più tardi.
La malattia di Fabry si presenta spesso con sintomi aspecifici che possono essere lievi e impercettibili e viene comunemente ignorata o diagnosticata erroneamente, portando a una sottovalutazione della sua prevalenza. [4].
Quali sono le cause della malattia di Fabry?
La malattia di Fabry è causata da mutazione di alfa-galattosidasi A gene (GLA) mappato sul braccio lungo del cromosoma X [5]. Ce ne sono centinaia di diversi patogeno varianti della mutazione, con conseguenti sintomi diversi e gravità variabile [6]. Gli uomini con la malattia di Fabry trasmettono il gene a tutte le loro figlie ma a nessuno dei loro figli. Le donne con malattia di Fabry hanno una probabilità 50% di trasmettere il gene alle loro figlie o figli. Alcune donne e ragazze sono colpite gravemente come uomini e ragazzi a causa di a casuale Inattivazione del cromosoma X [7].
Gb3 è un intermedio nella via di degradazione del globoside (un glicosfingolipide), un componente importante di alcune membrane cellulari. il enzima l'alfa-galattosidasi A catalizza il scollatura (clivaggio) del galattosio terminale di Gb3. La mancanza dell'enzima alfa-galattosidasi A porta all'accumulo di Gb3 in vari tessuti, che porta alla morte cellulare. I siti clinicamente più significativi di accumulo di Gb3 sono vasi sanguigni pelle, cuore, nervie reni [8].
Quali sono le caratteristiche cliniche della malattia di Fabry?
I tratti caratteristici della malattia di Fabry sono:
- Grappoli di angiocheratomi (piccoli, rosso scuro papule)
- intolleranza al calore e ipoidrosi (ridotta sudorazione)
- Episodi di acroparestesia (neuropatico dolore alle mani e ai piedi) (acroparestesia), precipitato a causa di stress o temperature estreme
- coperto dalle nuvole cornea
- Perdita dell'udito [9]
- Sintomi addominali come dolore, nausea, vomito e diarrea o costipazione [10]
- Ricorrente febbre, stanchezza, problemi psicologici e sociali
- Complicanze gravi e pericolose per la vita tra cui danno renale, malattie cardiache e ictus [11].
Dermatologico dimostrazioni si verificano in più di 70% di pazienti con malattia di Fabry, con a metà età di esordio a 17 anni [3].
Angiocheratomi
Gli angiocheratomi sono vasi sanguigni dilatati nella parte superiore dermapresentandosi come papule rosse o nere. Le papule no sbiancare con pressione e più vecchio lesioni Può essere verrucoso. grappoli o diffondere Gli angiocheratomi che compaiono per la prima volta nei giovani adulti dovrebbero avvisare i medici di una possibile diagnosi della malattia di Fabry.
Nella malattia di Fabry, gli angiocheratomi sono causati dall'accumulo di Gb3 nel dermico endoteliale cellule, portando al rigonfiamento e all'incompetenza delle pareti dei vasi [12]. Gli angiocheratomi si trovano solitamente nella zona del costume da bagno (dal ombelico alla parte superiore delle cosce, compresi i genitali). Anche più di un terzo dei pazienti sviluppare angiocheratomi e teleangectasia sulle labbra e all'interno della bocca [13].
Non vi è alcuna correlazione tra la gravità della malattia e l'estensione degli angiocheratomi. [14].
ipoidrosi
L'ipoidrosi è una caratteristica comune della malattia di Fabry che provoca pelle secca e intolleranza al calore. [quindici]. Probabilmente è il risultato di un'anomalia autonomo nervo risposta per deposizione Gb3.
Altro cutaneo manifestazioni della malattia di Fabry
La malattia di Fabry è identificata da altri segni cutanei, come:
- Linfedema della parte inferiore delle gambe [14]
- Viso spargolo e fine capelli [14]
- Il fenomeno di Raynaud [15].
Quali sono le complicanze della malattia di Fabry?
Le tre complicanze più gravi della malattia di Fabry sono malattie renali, malattie cardiache e cerebrovascolare malattia [3].
- La malattia renale varia dalla proteinuria isolata allo stadio terminale renale fallimento [3].
- Le malattie cardiache osservate insieme alla malattia di Fabry includono il ventricolo sinistro ipertrofia, insufficienza cardiaca, coronarica arteria malattia, e aritmie.
- La malattia cerebrovascolare include transitorio ischemico ictus e attacchi ischemici.
Come viene diagnosticata la malattia di Fabry?
I pazienti maschi e femmine vengono diagnosticati in modo diverso.
- I pazienti di sesso maschile possono essere diagnosticati attraverso un esame del sangue. Un livello molto basso di attività dell'alfa-galattosidasi A (<3%) es suficiente para diagnosticar la enfermedad de Fabry, mientras que un nivel superior al 35% excluye la enfermedad de Fabry. Los niveles de actividad entre estas dos cifras requieren un genetico esami per confermare la diagnosi.
- Le pazienti di sesso femminile hanno attività enzimatiche variabili con una percentuale significativa che mostra un'attività normale, pertanto la diagnosi della malattia di Fabry deve essere effettuata mediante analisi genetica.
- Se l'analisi genetica non identifica una specifica mutazione che causa la malattia per la malattia di Fabry, biopsia degli organi interessati (compresa la biopsia cutanea); questo può mostrare intracellulare accumulo di Gb3 [3,14].
Anche i familiari del paziente diagnosticato devono essere sottoposti a screening per la malattia di Fabry.
Analisi delle urine e ECG dovrebbe essere fatto per lo screening di malattie renali e cardiache anomalie.
Qual è diagnosi differenziale per la malattia di Fabry?
A causa della sua rarità e dell'ampia gamma di caratteristiche cliniche non specifiche, la malattia di Fabry è spesso mal diagnosticata e colloquialmente considerata una condizione di "grande impostore". [16].
Gli angiocheratomi non sono caratteristici della malattia di Fabry poiché lesioni solitarie possono verificarsi in individui sani. Si trovano anche in pazienti con ereditario emorragico teleangiectasia e negli altri disturbi lisosomiali, tra cui:
- Morbo di Schindler (una rara malattia metabolica ereditaria)
- Fucosidosi (un raro disturbo da accumulo lisosomiale che provoca un accumulo di zuccheri pericoloso per la vita)
- Aspartilglucosaminuria (un disturbo derivante da un difetto dell'enzima aspartilglucosaminidasi)
- Beta-mannosidosi (un disturbo risultante dalla ridotta attività dell'enzima beta-mannosidasi)
- Sialidosi (un disordine metabolico causato da una carenza dell'enzima neuraminidasi) [12,14].
Le diagnosi errate più comuni sono le seguenti:
- Reumatologico condizioni come reumatico febbre o dermatomiosite - in questi casi, le manifestazioni dermatologiche sono diverse nell'aspetto e nel corso del tempo
- malattie neuropsicologiche e fibromialgia - in cui vi è un'assenza di coinvolgimento di cuore, reni, nervi e pelle
- Policitemia vero ed essenziale trombocitemia - La malattia di Fabry non provoca un aumento piastrina raccontare
- Telangiectasia emorragica ereditaria: la malattia di Fabry di solito non causa epistassi o sanguinamento gastrointestinale
- Colon irritabile sindrome - questo non ha altre manifestazioni sistemiche [4,17].
Qual è il trattamento per la malattia di Fabry?
Una volta diagnosticata, ai pazienti maschi con malattia di Fabry viene spesso prescritta una terapia enzimatica sostitutiva, indipendentemente dalle caratteristiche cliniche. [9,14,18]. Le pazienti di sesso femminile devono essere trattate solo se presentano manifestazioni cliniche significative.
La terapia enzimatica sostitutiva fornisce ai pazienti l'enzima carente alfa-galattosidasi A. Riduce la gravità e la progressione delle manifestazioni della malattia, in particolare il dolore neuropatico e le malattie renali, riducendo la deposizione tissutale di Gb3. L'effetto della terapia enzimatica sostitutiva sulle manifestazioni cardiovascolari è meno chiaro [18].
I pazienti con malattia di Fabry possono richiedere il contributo di diverse specialità, inclusi medici di famiglia, cardiologi, nefrologi, neurologi e dermatologi per gestire le proprie manifestazioni organiche individuali. Il dolore neuropatico della mano e dei piedi può rispondere anticonvulsivanti, come la carbamazepina [18,19].
Il trattamento degli angiocheratomi include crioterapia, elettrocoagulazione, chirurgia di escissione e Essere terapia [12,14,20].
Qual è l'esito della malattia di Fabry?
La malattia di Fabry lo è progressivo e generalmente si traduce in una più breve aspettativa di vita. il previsione varia da paziente a paziente [21]. La sopravvivenza è significativamente ridotta nei pazienti maschi con forme gravi di malattia di Fabry a meno di 60 anni, spesso a 40 o 50 anni. La maggior parte dei decessi è dovuta a malattie cardiovascolari, inclusi ictus, malattie cardiovascolari, attacco di cuoree insufficienza cardiaca. morte per insufficienza renale, setticemia, e sono stati segnalati anche suicidi [18]. Il beneficio della terapia enzimatica sostitutiva sulla prognosi generale non è chiaro a causa dell'attuale mancanza di prove [18,22,23].